About Us
The Center for Immunotherapy and Precision Immuno-Oncology (CITI) Computational Oncology Platform group uses various core technologies for processing and analyzing data based on high-throughput next-generation sequencing from the raw data to ready-to-use results. Through partnerships with scientists, clinicians and industry, CITI accelerates the development and approval of novel immunotherapy treatments and applications. Our partners benefit from early access to scientific advances, expansive clinical-genomic datasets and expert advice and analysis.
Our scientific & technology areas include:
- Mutation Calling
- Neoantigen Analysis
- Copy-Number Analysis
- Tumor Clonality Analysis
- Gene Expression Analysis
- Immune Infiltration and Immune Activity Analysis
- Differentially Expressed Gene and Pathway Analysis
- Single Cell RNA Sequencing
Learn more about our scientific & technology areas.
Services
The Computational Oncology Platform offers bioinformatics & computational biology support as a core service to investigators. Learn more about our services. To view prices and request services, visit our iLab page.
Contact Us
Interested in learning more? Contact us today at [email protected].
Our Team

Vladimir Makarov, MD, MS
Staff Scientist
Dr. Vlad Makarov is the Scientific Director of the Computational Immunology Platform (CIP), a subdivision of CITI’s Precision Oncology Program. Since joining the Cleveland Clinic in 2020, Dr. Makarov and his team have launched bioinformatic computational fee-for-project shared resource services for analysis of high throughput DNA/RNA/single cell RNA-sequencing data output. Dr. Makarov earned his MD in St. Petersburg, Russia. His background in population genetics, cancer genomics, immuno-oncology, and software development has poised him to lead a team of computational genomic data scientists. He has been involved in various genomic consortium projects and has led the development and optimization of variant-calling pipelines for whole genome (exome and transcriptome) analyses. He has also co-authored three published genome analysis tools: AnnTools, RDXplorer, and NAseek.










Salendra Singh, MS
Software Developer
[email protected]
Salendra Singh is a software developer and data scientist on CITI’s computational immunology team. He obtained his Master of Science in Biochemistry and Molecular Biology from Georgetown University and has been working in the field of computational biology for over 11 years. He most recently served as a Senior Bioinformatics Scientist for the Case Comprehensive Cancer Center. At CITI, Salendra plays a leading role in the development and management of departmental web resources while also continuing research efforts in immunotherapy, genomics, oncology, and biomedical sciences. His research focuses on developing systems biology tools and contributing to integrative genomics, digital pathology, radio genomics, and single cell/nuclei and spatial genomics. Find Salendra on LinkedIn, Google Scholar, and Twitter.
Analyses & Technology
The Center for Immunotherapy and Immuno-Oncology (CITI) Computational Biology Division utilizes large-scale technologies and high-throughput profiling for collaborative projects. These analyses are also offered as core services to support research community. Through partnerships with scientists, clinicians, and industry collaborators, CITI computational faculty focus state-of-the-art computational biology and NGS-based discovery. Our partners benefit from early access to scientific advances, expansive clinical-genomic datasets, and expert advice and analysis.
The web-based immunogenomics analysis platform developed by CITI, IOExplorer, allows collaborators to easily visualize their results, explore analytic variations, and compare their cohorts with published datasets.
The CITI computational division, in collaboration with Cleveland Clinic’s Pathology & Laboratory Medicine Institute (PLMI) and the Taussig Cancer Institute (TCI), operates the Precision Oncology Program. As part of this program, we support CCF cBioPortal, which provides easy access to genomic data from CCF patients in a scalable, mineable, easy to use portal environment linked with treatment recommendations.
Below are some of our primary pipelines and services.
Having been a key early member of the TCGA analysis team, our group has extensive experience with the analysis of cancer genomes. Our mutation calling pipeline processes whole-exome, whole genome, and targeted gene sequencing panels to identify somatic and germline mutations. Illustrative example analyses are shown below.
Mutation calling pipelines are fully automated following the best practices recommended by the National Institutes of Health.
Shown below is an example of genomic output for a recent clinical trial.
 - Variant Calling 10.13.21.jpg)
Representative integrated mutational data from melanoma. Data types are labeled above. Riaz et al, Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab, Cell, 2017 Nov 2;171(4):934-949
Neoantigens are mutated peptides that form the basis of how immune cells recognize cancer cells. Our research group was the first to show that tumor mutations and neoantigen burden drive immunotherapy treatment benefit in patients, a finding that is a cornerstone of the understanding of immunotherapy’s mechanism of action. We can use computational methods to predict neoantigens using various methods and incorporate the predictions into integrated genomic analyses. By using concurrent HLA genotyping, we can also characterize HLA divergence (HED) and immunopeptidome content and use this information to elucidate cancer cell epitope presentation.
 - NeoAntigen 10.13.21.jpg)
Neoantigen formation. Different types of mutations can form neoantigens in cancer cells that are recognized by the immune system.
 - NeoAntigen 10.13.21.jpg)
HLA evolutionary divergence predicts for immunotherapy efficacy in melanoma patients treated with immune checkpoint blockade.
Allele-specific copy-number analysis is performed using DNA sequencing data. The fraction of the copy-number-altered genome is defined as the fraction of the genome with either non-diploid copy-number or evidence of loss of heterozygosity. Sample purity and ploidy are also estimated for use in downstream analysis. This is helpful to characterize copy number alterations in normal and disease states.
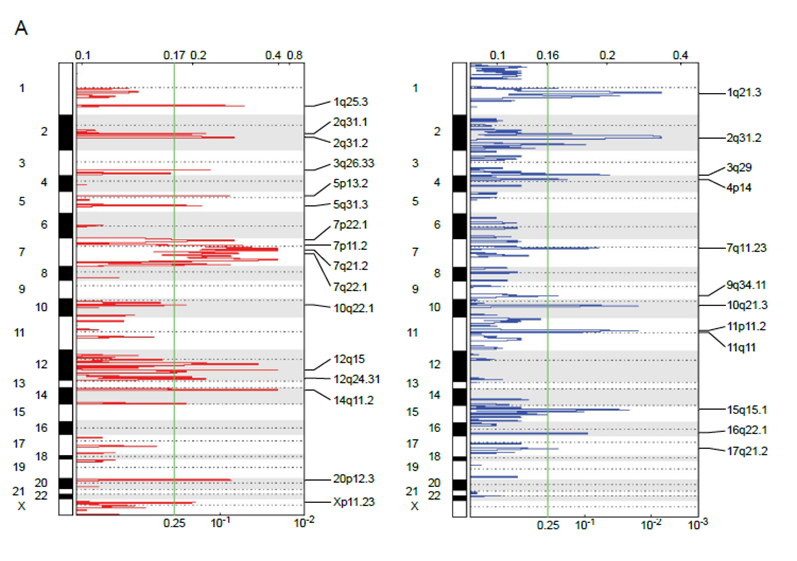
Example of copy number analysis identifying genomic changes in cancer. Ganly et al. Cancer Cell 2018.
Cancer cell fraction (CCF) represents an estimate of the fraction of cancer cells carrying a given mutation. Analysis of CCF can help identify cell subclones that independently develop over the lifetime of a tumor and estimate their relative fitness and susceptibility to immune targeting. For each mutation, we calculate the CCF based on variant allele frequency, copy number, and sample purity estimated in previous steps. Furthermore, we classify single nucleotide variants (SNVs) into clonal and sub-clonal variants depending on the confidence interval (CI) of the CCF estimation. SNVs for which the lower bound of the CI exceeded 95% are considered clonal mutations, others sub-clonal.
Bulk RNA-seq experiments measure the average expression of each gene across an entire transcriptome. Reads obtained by the experiment are aligned to the latest build of the human or model organism genome. Raw gene-level count values are normalized by sample specific size factor and FPKM (Fragments Per Kilobase Million) values are reported. The normalized values are used to find significantly different expression between specified groups of samples. Canonical pathway analysis of differentially expressed genes is then performed by pathway analysis software. Tools like GSEA and network analyses are used to define the biological pathways of altered transcriptional programs. Additionally, more advanced analyses can be performed from bulk RNA experiments, such as gene fusion expression and TCR clonality. We have recently added the Immunarch package to further analyze T-cell receptor (TCR) and B-cell receptor (BCR) repertoires.
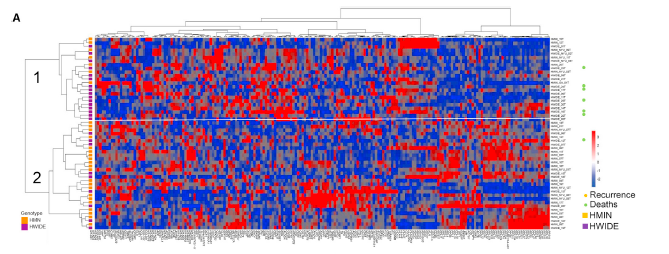
Example of unsupervised clustering analysis of gene expression data from thyroid cancers.
In mixed cell populations, bulk RNA-seq experiments lack the resolution required to identify the cell types responsible for altering gene expression between groups. We deploy high resolution single cell RNA-seq (scRNA-seq) to gain a better understanding of how individual cell populations are altered during therapeutic response. In scRNA-seq experiments, reads are tracked to individual cells and used to construct expression profiles, enabling accurate detection of different cell types. ScRNA-seq using 10X genomics 3’ and 5’ protocols with VDJ and Feature library preparations are used in the laboratory with both tumor and PBMC samples. Downstream processing includes the standard 10X genomics Cell Ranger 6.0.0 software pipeline, followed by additional quality control checks, data normalization, and batch correction using the R package Seurat v4.0. These methods have been validated on multiple immunotherapy studies including on renal cell carcinoma and breast cancer. With verified cell signatures and reference datasets provided from Seurat, we can confidently identify immune cell types to identify changes under immunotherapy, including therapeutic resistance.
 - Single Cell RNA Sequencing 10.13.21.jpg)
Mutational signatures are patterns of nucleotide alterations in tumor genomes that are characteristic of various mutational processes, including carcinogenic insult, aging, and DNA repair defects. Our mutational signatures computational pipeline utilizes mathematical methods to estimate, from NGS samples, the contribution of various known mutational signatures. The pipeline includes mutation calling, tri-nucleotide context matrix generation and normalization, negative matrix factorization, non-negative least squares regression, prediction of mutational signatures, and transcriptional strand-based mutational signature analysis (reference).
Immunogenomics
To support research and clinical operations, CITI collaborates with multiple partners within and beyond Cleveland Clinic to develop computational systems that facilitate the efficient utilization of our extensive data collections. The basic components of our core immunogenomics data systems are: multiple sources of clinical and genomic data, a central data warehouse where the data are integrated (MaGiC), and applications to access and use the data, such as cBioPortal and IOExplorer.
CITI works closely with collaborators from multiple Cleveland Clinic divisions, including the Pathology and Laboratory Medicine Institute (PLMI), the Taussig Cancer Institute (TCI), the Enterprise Analytics (EA) division, and departments within the Lerner Research Institute (LRI). Through this joint effort, we integrate disparate data sources into a unified precision oncology data warehouse called the Molecular and Genomics Integrated at Cleveland Clinic (MaGiC) database. Data sources include databases and files overseen by PLMI, TCI, and EA, which are hosted on various data systems such as Teradata, MS SQL Server, Oracle, and flat files. Key types of data include sample and diagnostic, such as cancer types, staging, and procedure dates; molecular, such as raw sequencing results and mutation calls from various panels and providers such as Tempus and Caris; and curated treatment and outcome data. After integration, anonymized data extracts are provisioned for exploratory analysis to client applications such as cBioPortal and IOExplorer, or for advanced analysis directly to IRB authorized clinicians or researchers. MaGiC is intended and designed to facilitate future expansion to any disease type.
cBioPortal is an open source, interactive graphical cancer genomics web app developed in association with Memorial Sloan Kettering Cancer Center and used by major cancer centers. CITI and our partners are providing cBioPortal to the Cleveland Clinic community in support of both clinical and research uses. The cBioPortal includes, in addition to public datasets such as TCGA, genomics data from major gene panels used at CCF, such as Tempus and Caris, and will expand to include all CCF panels and other genomics data. In addition to genomics data, cBioPortal contains associated clinical data such as diagnoses, treatments, and outcomes. Access is restricted according to IRB and clinical authorization. For additional details about cBioPortal functionality, please see https://www.cbioportal.org/tutorials.
MAGIC and cBIOPortal are key elements that support precision medicine in research and clinical activities. They are integrated with the CCF cancer center genomics tumor board. The system provides genomics reporting for ordering clinicians and enables scalable and minable genomics research for clinical trials.
The pace of cancer immunotherapy research is accelerating, increasing the volume of data available for developing novel therapies, discovering and refining biomarkers for more precise targeting of existing therapies, and making other advances. But utilizing existing data to formulate new hypotheses, address clinical questions, etc. continues to require extensive bioinformatics expertise and time, which is an impediment to the efficiency, scope, and pace of research.
While powerful, cBioPortal (above) is not designed to specifically investigate immuno-oncology datasets and thus does not provide certain functionality key to this field of research, such as analysis of HLA types and diversity. To fill this gap and make exploratory IO analyses more accessible, rapid, and powerful, we have developed an IO-specific interactive graphical web application, called IOExplorer.
Key features of IOExplorer include:
- An interactive graphical user interface with convenience features, such as point-and-drag data selection and data type tagging.
- Meta-analysis ready datasets and features.
- Pre-/on-treatment model for samples.
- Multiple analysis modules: distribution, correlation, mutation, expression, volcano (beta), HLA, survival.
- Analyses can be saved, resumed, and shared.
- Interactive tutorials and contextual help.
- Collaborative development with researchers and clinicians.
Datasets & Standardization
The first obstacles to exploratory and meta-analysis are simple in concept but complicated to implement: acquiring and standardizing available datasets. Whereas acquisition is a bureaucratic exercise, meaningful analyses that pool or compare data between or across studies require that the data be standardized. Effective standardization demands extensive technical expertise and computing resources. IOExplorer includes key published IO datasets (see below) that we have acquired and standardized by reprocessing the raw data through our established pipelines (where permitted by data use agreements ) (see above). In addition, IOExplorer includes features to facilitate analyses of multiple datasets, including capabilities to:
- Pool and filter data according to any user-specified criteria. Filtering criteria are specified via analysis modules, either by predefined groupings such as quartiles, or visually by click-and-drag selection.
- Correct expression data in real-time across studies or batches.
- Use built-in standardized clinical attributes (e.g., response to therapy).
- Analyze cohorts created from pooled data or compare between cohorts.
 - Datasets 10.13.21.jpg)
The following datasets are currently included in IOExplorer (as of December 2021) and we frequently update as new studies are published, processed, and standardized. In general, data use agreements restrict use to analyses within IOExplorer, not redistribution of original data.
- Snyder et al., NEJM 2014
- Riaz et al., Cell 2015
- Rizvi et al., Science 2015
- Van Allen et al., Science 2015
- Gao et al., Cell 2016
- Hugo et al., Cell 2016
- Ott et al., Nature 2017
- Roh et al., Sci. Transl. Med. 2017
- Auslander et al., Nat. Med. 2018
- Cristescu et al., Science 2018
- McDermott et al., Nat. Med. 2018
- Miao et al., Nat. Gen. 2018
- Miao et al., Science 2018
- Kim et al., Nat. Med. 2018
- Cloughesy et al., Nat. Med. 2019
- Gide et al., Cancer Cell 2019
- Liu et al., Nat. Med. 2019
- Zhao et al., Nat. Med. 2019
- Valero et al., Nat. Gen. 2021
Analysis Modules
Distribution
Exploratory data analysis is an essential first step of rigorous statistical analysis. It involves an examination of basic features of a dataset to understand the characteristics of its data points (e.g., samples) and its relationship to other datasets, helping to define the applicability of various statistical models and tests, etc. IOExplorer provides two analysis modules dedicated to examining the characteristics of individual variables (the Distribution module) or pairs of variables (the Correlation module). As with all modules, analyses may be confined to a single dataset or applied to cohorts consisting of combinations of 1-to-N datasets and 0-to-N filters. In addition to these to purpose-built modules, all IOExplorer modules provide functionality for exploratory for specific attributes relevant to immuno-oncology.
 - Datasets 10.13.21.jpg)
 - Datasets 10.13.21.jpg)
Mutation
Mutation analysis is a cornerstone of immuno-oncology. Loss-of-function mutations in tumor suppressor genes and gain-of-function mutations in proto-oncogenes are associated with specific cancer histologies and may be predictive of specific treatment outcomes and thus of high clinical relevance. Furthermore, the overall mutation load of a tumor (TMB) is generally predictive of the effectiveness of immunotherapy: high TMB is associated with improved immunotherapy outcomes in many patients, possibly via generating effective neoantigen targets for immune system activity. IOExplorer permits the user to select one or more predefined gene sets relevant to immunotherapy (e.g., CD8+ T-cells), or to enter any gene(s) of interest. Single and short nucleotide variants are displayed on oncoprint displays with customizable axis, ordering and scaling, permitting the user to visualize associations between gene sets and any clinical or genomic variable(s) of interest. Copy-number variation analysis is under development.
 - Datasets 10.13.21.jpg)
Expression
Analysis of altered tumor expression patterns is another cornerstone of immunogenomics and goes hand in glove with mutation analysis. Characteristic histologies and predicted treatment outcomes may be associated with under-expression of tumor suppressor genes or over-expression of oncogenes, which may be associated with detected mutations or mutations to regulatory regions not included in gene sequencing panels or by more complex genomic alterations.
RNA-seq results are notoriously sensitive to batch effects, making it particularly challenging to implement general comparisons between or pooling among batches or studies. IOExplorer implements two general approaches to expression data normalization. First, all studies include user-selectable options for both raw and customary per-study normalization (VST, TPM, CPM). Furthermore, IOExplorer provides capabilities for real-time cross-study batch correction, which is currently limited to user selected pairs of studies (Volcano module), but with more generalized real-time normalization under development.
As with mutations, IOExplorer provides the user to interactively explore patterns of altered expression in tumor samples using predefined or custom gene sets, displayed as heatmaps, and to associate altered expression patterns with user-configurable clinical and molecular variables.
 - Datasets 10.13.21.jpg)
 - Datasets 10.13.21.jpg)
HLA Divergence
If mutation and expression go hand-in-glove, then HLA is the hand. HLA-I is key in determining which (if any) antigens are displayed on the surface of tumor cells for immune recognition. Until recently, the capacity of cells to display neoantigens was typically analyzed by simply examining whether HLA-A, HLA-B, and HLA-C were homozygous or heterozygous. Such an approach ignores the great variability between different allele pairs, some of which are nearly identical and others of which are highly diverged. Members of our team developed an improved approach to characterizing HLA diversity based on the physiochemical differences between heterozygous alleles, called HLA evolutionary divergence (HED) (reference). IOExplorer provides multiple interactive displays for both HLA-I heterozygosity and HED of cohorts, making it an extremely powerful tool for HLA analyses. We have created and maintain the HED R package which has been updated to the latest Release 3.46 of IEDB (Oct 2021)
 - Datasets 10.13.21.jpg)
 - Datasets 10.13.21.jpg)
Survival
Finally, IOExplorer endpoint analysis is currently provided as Kaplan-Meier overall survival plots with configurable Wilcoxon weighting, optional confidence interval display, and statistics.
 - Datasets 10.13.21.jpg)
Services
Bioinformatics & Computational Biology Services
The Center for Immunotherapy and Precision Immuno-Oncology (CITI) Computational Oncology Platform offers bioinformatics support as a core service. We use industry standard tools for data processing and analysis. Having been original core members of the Cancer Genome Atlas (TCGA) team, our pipelines are extensively benchmarked and based on best practices. Users are encouraged to include the description to their papers to support the methods sections within their manuscripts. All deliverables are in industry standard formats such as BAM, VCF or MAF. Quality control (QC) metrics are also provided, allowing investigators to gauge the quality of sequencing in a unified manner. We encourage researches to schedule a meeting with us prior to data processing/analysis.
The CITI computational team currently offers services for the following data types:
- STAR alignment to reference genome, Pre/Post alignment QC collection, raw and normalized read counts over genes.
- Differential gene expression as specified by users. Users provide metadata files according to provided templates.
- Gene set enrichment analysis (GSEA) and pathway analysis and xCell deconvolution for 64 immune cell types.
- Alignment to reference and Pre/Post alignment QC collection.
- DNA profiling (genetic fingerprinting). Optional, add to post-alignment QC.
- Post-alignment WES/WGS data processing.
- Somatic or germline variant calling and annotation (SNV/INDELS).
- Allele Specific Copy Number calling.
- Microsatellite instability (MSI) status.
- Standard Cell Ranger pipeline, Loupe objects per sample and per project, Pre/Post alignment QC metrics collection.
- Post-alignment Single Cell RNASeq analysis. Includes standard Seurat pipeline, generating Integrated Seurat Object and automated cell typing.
- T-cell/Antibody Immune Repertoires Analysis. Data normalization, CDR3 identification, clonal dynamics, and repertoire features such as diversity and Shannon metrics. We will need to discuss metadata, comparison, and other parameters prior to analysis. For TCR Sequencing, please see the Discovery Lab.
- Uploading genomics data to dbGAP or other data repositories for publication.
- Additional custom analysis may be possible for additional fees, please contact us for availability. Services beyond the basic analyses provided above will be available based on feasibility and availability.
Researchers interested in continuing working with us through more extensive collaborative efforts after basic data processing/analyses has been completed may contact us today.
Future Services
Work in Progress coming soon
We are currently in the process of adding the new pipelines.
ChIP-Seq
ATACseq