Our Laboratories
The Center for Immunotherapy & Precision Immuno-Oncology brings together researchers from multiple disciplines to develop innovative immune system-based personalized cancer treatments utilizing genomic analysis and high-throughput immunoprofiling.
Our department includes research laboratories, shared research platforms, developmental therapeutics and precision medicine programs.

What is Immunogenomics?
Cancer is complex. Cancer cells become deviant, growing and functioning abnormally. They cause disease symptoms and sabotage the function of the organs they invade. Our immune response to cancer is equally complex. When cancer turns fatal, it often means the immune system is no longer able to hold back tumor growth.
Our researchers and clinicians are using immunogenomics—the combined parallel study of the genomics of tumor and immune cells—to better understand the body's immune response to cancer.
Unfortunately, immunotherapies, which boost the strength and specificity of immune cells trained to fight disease, are not currently universally effective. Immunotherapies work very well on some types of cancer, but not others—and very well in some patients, but not others. Our team is hard at work to understand why this is and how to make immunotherapies effective for every patient.
Other questions we aim to answer are: How does immunotherapy change the immune cell repertoire, and thus the selective pressure leveled against tumor cells? How does the tumor cell population evolve as this pressure changes? What is going on inside a tumor when anti-tumor immunity fails?
Ultimately, we seek to uncover how to tailor immunotherapy so that every patient experiences the greatest possible benefit with the least risk. We are working to develop new immunotherapy strategies to improve outcomes.
Principles of Immunotherapy & Immunogenetics
In cancer, normal cells lose the ability to control their own growth and become immortal. While this can sometimes occur as a result of rare inherited mutations in critical genes, these pro-cancer cell changes more often occur gradually as a cell sustains damage to its DNA over time. DNA serves as a blueprint for how a cell should function, so changes in DNA, called mutations, can be dangerous.
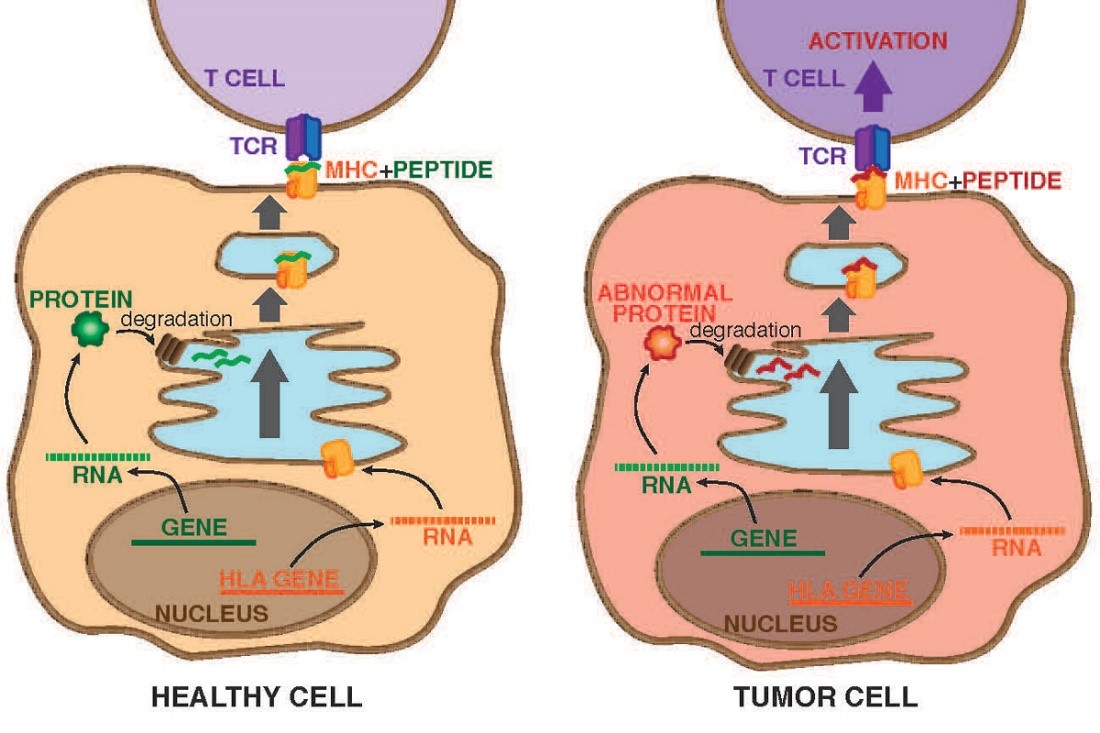
In addition to fighting off infections due to viruses and bacteria, our immune system also surveils our body to recognize and protect us from the dangerous effects of cancer cells. Both mutations that help cancer cells grow ("driver mutations") and those that do not ("passenger mutations") can cause cells to express new versions of native proteins. Because these proteins do not occur anywhere else in the body, the immune system can recognize them as foreign. These foreign proteins, recognizable by our immune system, are called neo-antigens. The key immune cells that protect us from cancer by recognizing these neo-antigens are called T cells.
Cancers evolve through characteristic interactions with the immune system, referred to as the "three Es"—elimination, equilibrium and escape. Elimination occurs when the immune system destroys an abnormal growth before it becomes a cancer. Equilibrium refers to scenarios when the immune system keeps the tumor from growing but cannot eliminate it completely. Lastly, when the immune system is either suppressed by the tumor or no longer recognizes it as foreign, evasion of cancer-causing cells occurs, also known as immune “escape.”
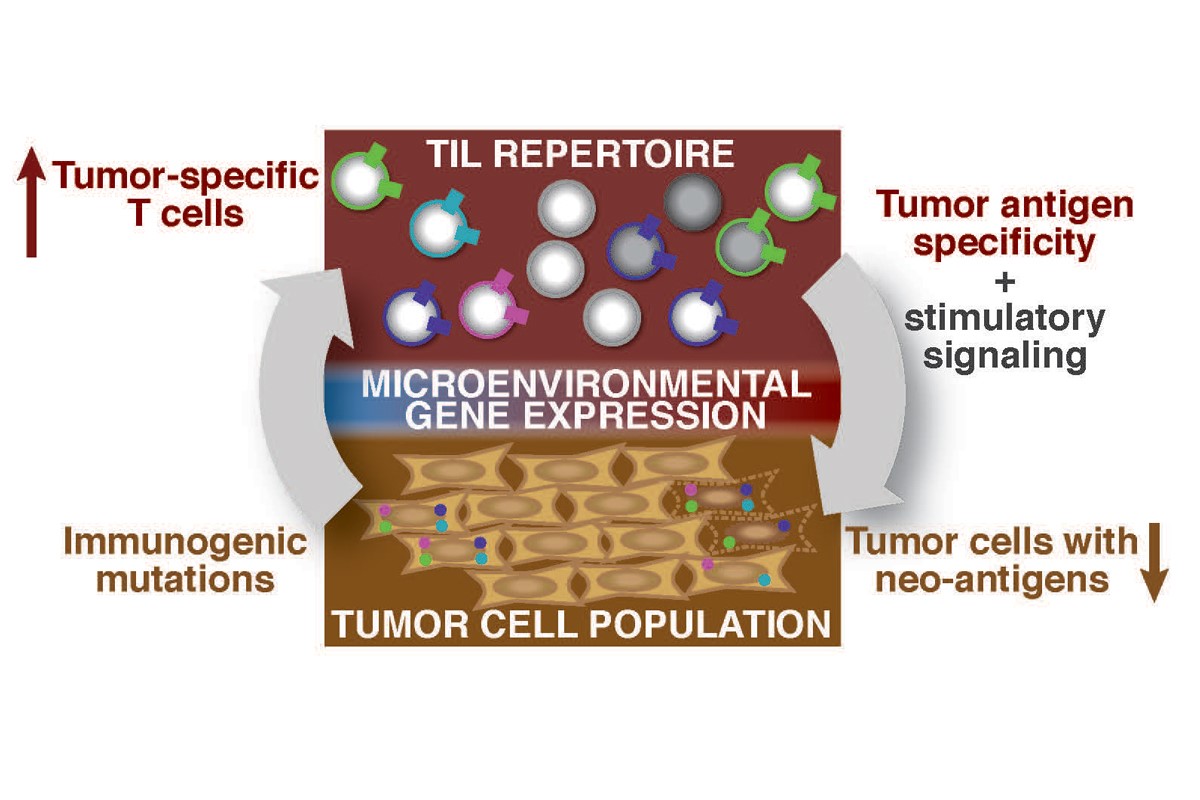
Immunotherapies reinvigorate the immune system when a tumor is escaping, helping to restore equilibrium or even eliminate the tumor. Immunogenomics can provide information about the landscape of mutations and neo-antigens in a tumor as it changes over time (including during the course of therapy), helping us to better understand the biological mechanisms that make immunotherapy successful. In parallel, immunogenomics can also help us understand the specificity and strength of the immune response as it acts upon the tumor. Research in this area will help elucidate how a cancer escapes immune control and inform the development of better therapies for patients.
Cancer is an evolutionary process and tumor cells can accumulate hundreds of mutations as they grow and divide. Some of these mutations are immunogenic—recognizable as "non-self" by our immune system. For a mutation to be immunogenic, the mutated protein has to be processed inside the cancer cell, and the resulting mutated peptide (called a neo-peptide) must bind to one of the patient's major histocompatibility complex (MHC) class I molecules in order to be presented on the cell surface. Then, a T cell must be able to recognize the neo-peptide with its T cell receptor (TCR) in order to subsequently trigger an immune response. The physical feature that helps the TCR to recognize the neo-peptide is called an epitope. Increasing evidence suggests that the immune response to these mutation-derived antigens is very specific and critical for a successful response to immunotherapies, including immune checkpoint blockade and adoptive T cell therapy. (For background and research, see the following additional references: 1, 2, 3, 4, 5, 6, 7.)
When a T cell recognizes an antigen with its T cell receptor, it activates, or "turns on," and begins to proliferate. Once the number of activated T cells expressing that specific receptor increases, they can work together to kill the invading tumor cells or any other threats that may arise.
Any immune reaction takes energy to sustain, which can cause damage to healthy tissues and hinder the body's ability to react to other challenges. Importantly, however, the immune system has mechanisms to maintain balance.
When a T cell becomes activated, it begins to express other receptors on its cell surface that serve as "off switches." These switches can be triggered by other cells (such as other immune cells, healthy tissue and even tumor cells) to shut down the killing action of the activated T cells. Furthermore, the longer a T cell is exposed to the antigen it recognizes, the weaker its ability to kill becomes—a phenomenon called exhaustion. It is thought that both of these mechanisms exist because most threats to the immune system are acute, which means they occur suddenly (like an infection) and are cured by the immune response in days or weeks. Tumors (as well as some infectious diseases), on the other hand, pose a chronic challenge in which the immune cells are stimulated by the same antigens over longer periods of time – this can occur over months, or even years. The T cells specific to that threat may become exhausted or actively suppressed by both tumor cells and healthy cells, triggering their "off switches" to protect themselves.
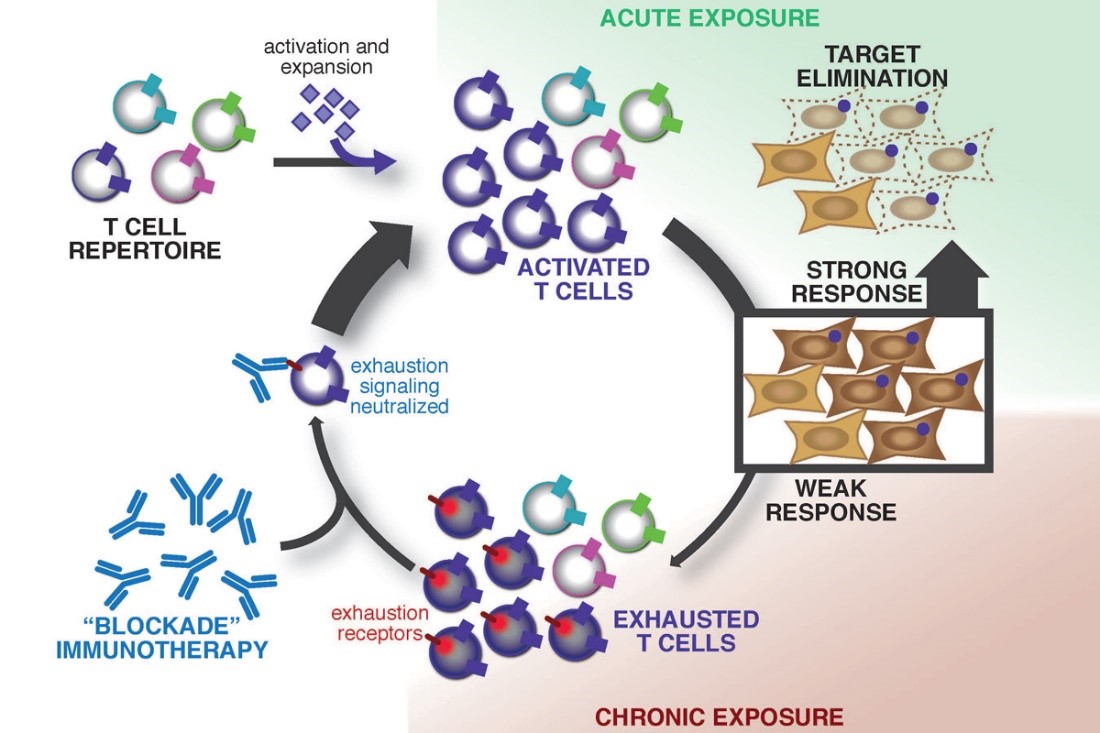
Many immunotherapies used against cancer are designed to protect or rescue T cells from this exhaustion or suppression, allowing tumor-specific immune cells to fully regain their killing functions. We use high-throughput sequencing of both the immune cells and the tumor cells to: 1) improve immunodiagnostics for determining what aspect of a patient's immune system is functioning suboptimally; 2) describe how the mutations in the tumor population change when selectively killed by rescued immune cells; 3) understand why these immunotherapies work better in some patients than in others; and 4) devise precision combinations of immunotherapies with chemotherapy and radiation therapy to maximize the killing of tumor cells while minimizing the damage to healthy tissues in every patient.
Core Technologies
Mutations accumulate in cells due to environmental insults, such as UV light and cigarette smoke, and form sporadic DNA replication errors that occur during normal cell proliferation. Mutations that confer the ability to proliferate unchecked by the body's normal regulatory systems are often referred to as driver mutations. Cells with driver mutations can become abundant in the tumor population. Every time these cells divide, there is a chance that additional mutations will occur due to DNA copying errors. Thus, in addition to driver mutations, tumor cells often accumulate random damage to many other parts of the genome, including those that do not accelerate cancer's growth (called passenger mutations).
The mutational landscape of a tumor is composed of both driver and passenger mutations, which can be identified using high-throughput next-generation sequencing. Studying the number of each, their abundance in the population, and which mutations seem to have evolved together can collectively reveal key information about the selective pressure the tumor is under (including competing for limited resources like nutrients and oxygen, struggling to maintain essential cell processes despite rapid growth, or being attacked by the immune system). Studying these factors can also inform precise combinations of therapies to target the genetic and immunogenic weaknesses of the tumor.
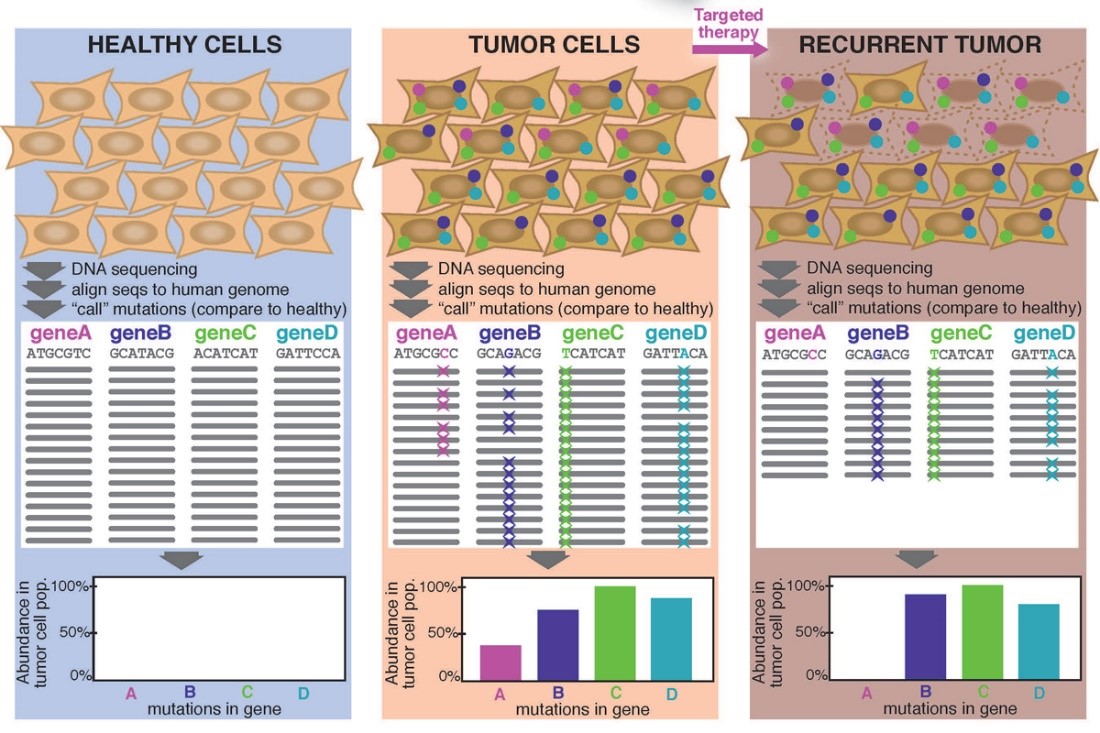
We use whole-exome sequencing, whole-genome sequencing and targeted gene sequencing to identify the genomic factors affecting antitumor immune activity. Our refined pipeline maps raw sequence reads to the human reference genome; annotates the positions of insertions, deletions, and nucleotide variations; and removes artifacts from library preparation.
Clonality
We are interested in understanding the clonal composition of tumors. A clone is a cluster of cells that shares the same mutations, possibly due to a shared lineage. When a tumor contains many shared lineages, it is called "subclonal." These distinctly arising subclones can accumulate new mutations that provide growth advantages, allowing them to out-grow less competitive subclones. Over time, the most competitive subclones make up a higher overall proportion of the tumor.
However, not all subclones in a tumor necessarily respond to immunotherapy the same way. Some subclones may carry mutations that cause a stronger immune response than others. Therefore, it is important to understand the clonal composition of tumors in order to design therapeutic strategies that target enough of the tumor to perturb its growth at a clinically measurable level.
We use genome sequencing to estimate the relative frequency of cells within a tumor that carry mutations. For each mutation, we calculate the cancer cell fraction (CCF) based on variant allele frequency of the mutation, its copy number, as well as the sample's purity. CCF analysis can help us to identify new tumor subclones that develop independently over the lifetime of a tumor. It can also help to deduce the relationship between the fitness of the new subclones relative to previous tumors, which can guide immune targeting strategies.
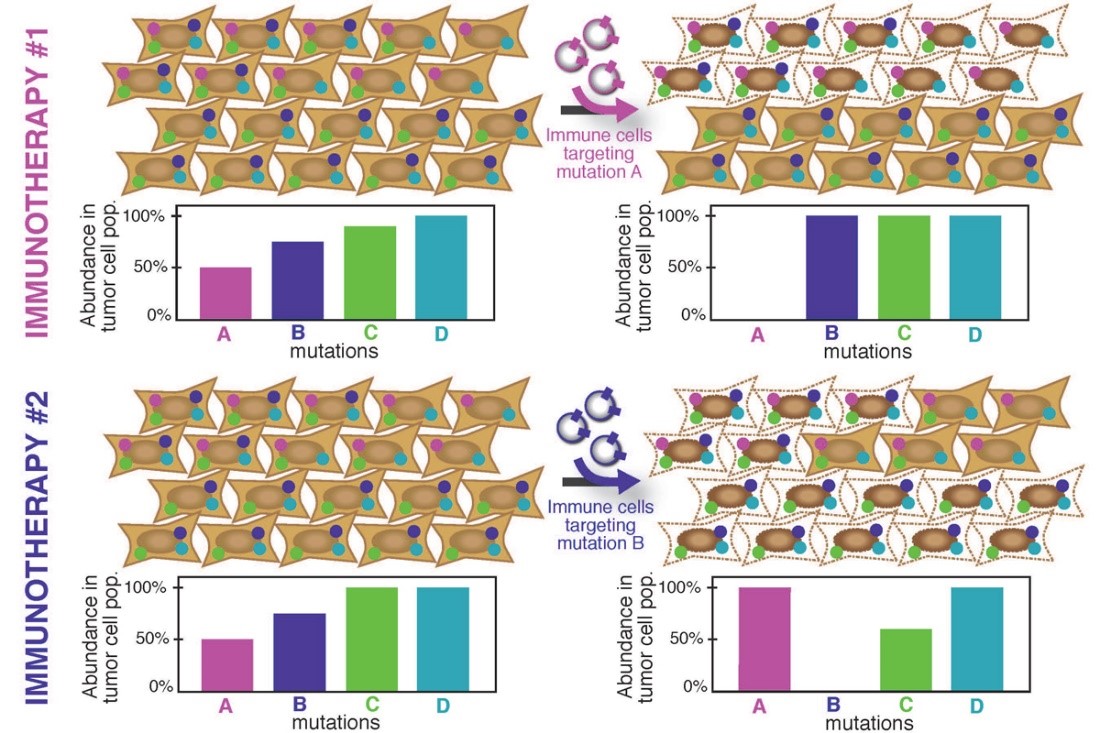
Neo-Antigen Prediction
A major obstacle to the development of a strong, effective immune response to a growing tumor is the fact that tumor cells are very similar to healthy tissue. Antigens that arise in tumor cells due to mutations (neo-antigens) allow the immune system to recognize those tumor cells as non-self and can thereby trigger a tumor-specific immune response. It is thought that the number of neo-antigens present in a tumor is a crucial factor in determining whether an immunotherapy will be successful at marshaling an effective antitumor immune response.
We are actively developing novel computational approaches to identify neo-antigens in human cancers. Our current method utilizes the same somatic mutation-calling pipeline as described above (see Genomic Sequencing), followed by neo-epitope analysis.
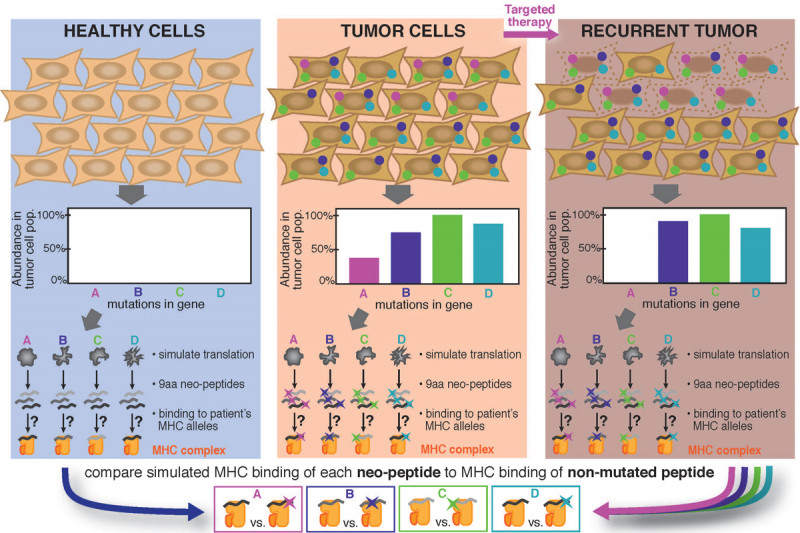
We are developing algorithms for predicting neo-epitopes. Typical algorithms translate all mutations identified by the genomic mutation pipeline, generate candidate peptides that would contain the mutations, and predict the ability of the peptides to bind to MHC molecules. Peptides for which the mutated version is more strongly presented than the wild type are considered potential neo-antigens.
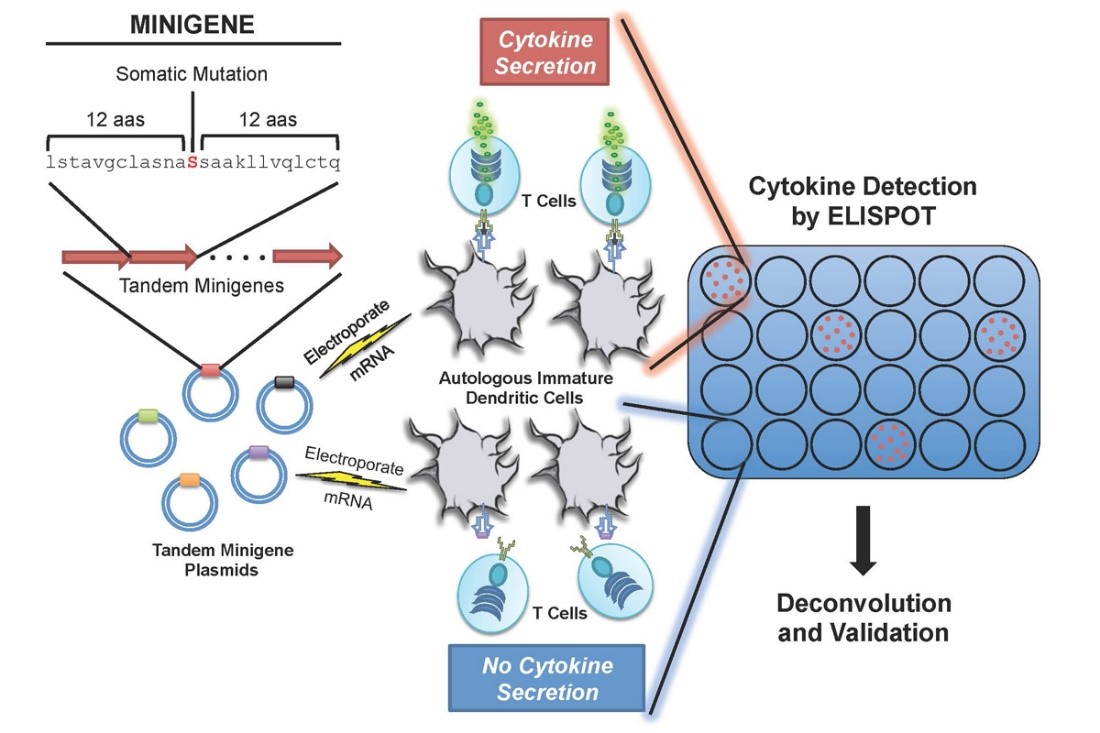
To better understand the specific interplay between a patient's mutations and the immune system, mutant peptides are systematically tested for immunogenicity—the ability to activate T cells taken from the same patient. Results of this type of antigen screening can help in the creation of more personalized immunotherapies, such as tumor-specific vaccines or adoptive T cell therapies. Furthermore, we seek to understand the relative contributions of different types of mutations and antigens to effective immune responses, with the ultimate goal to make patient-specific therapies more precise.
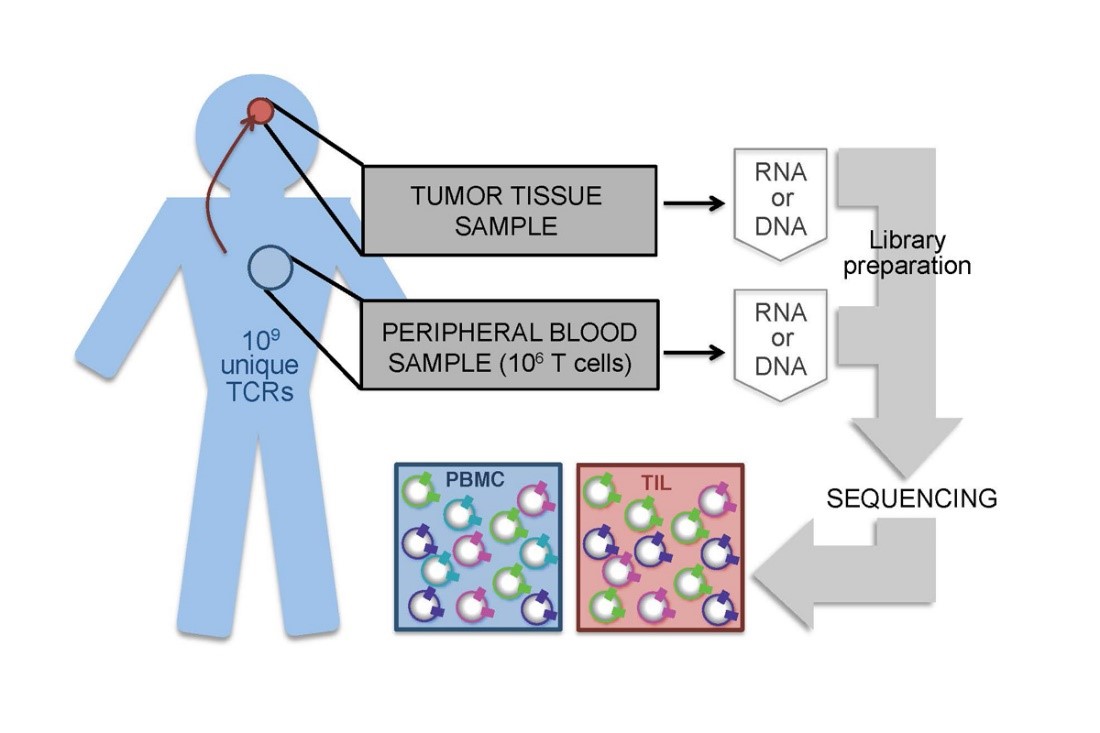
Adaptive immune cells (T cells and B cells) help us to recognize specific threats, such as microbial pathogens (e.g., bacteria, viruses, fungi) and tumors. Each T cell or B cell expresses a receptor on its surface—the T cell receptor (TCR) or B cell receptor (BCR), respectively—that can bind to a particular molecular target and differs from one immune cell to the next. When a TCR or BCR finds its target molecule (called an antigen) the T or B cell is signaled to divide and multiply. Each receptor is unique, generated by random DNA recombination and alteration during development into a mature T or B cell. The number of different TCRs that can be generated by one person is extensive – between 1012-1020 over the course of a lifetime, with about 109 present in the repertoire at any given time. It is the vast diversity of these receptors that both enables any one person to respond to antigens his or her immune system has never encountered before and to raise an "army" against a particular antigen if it represents a threat.
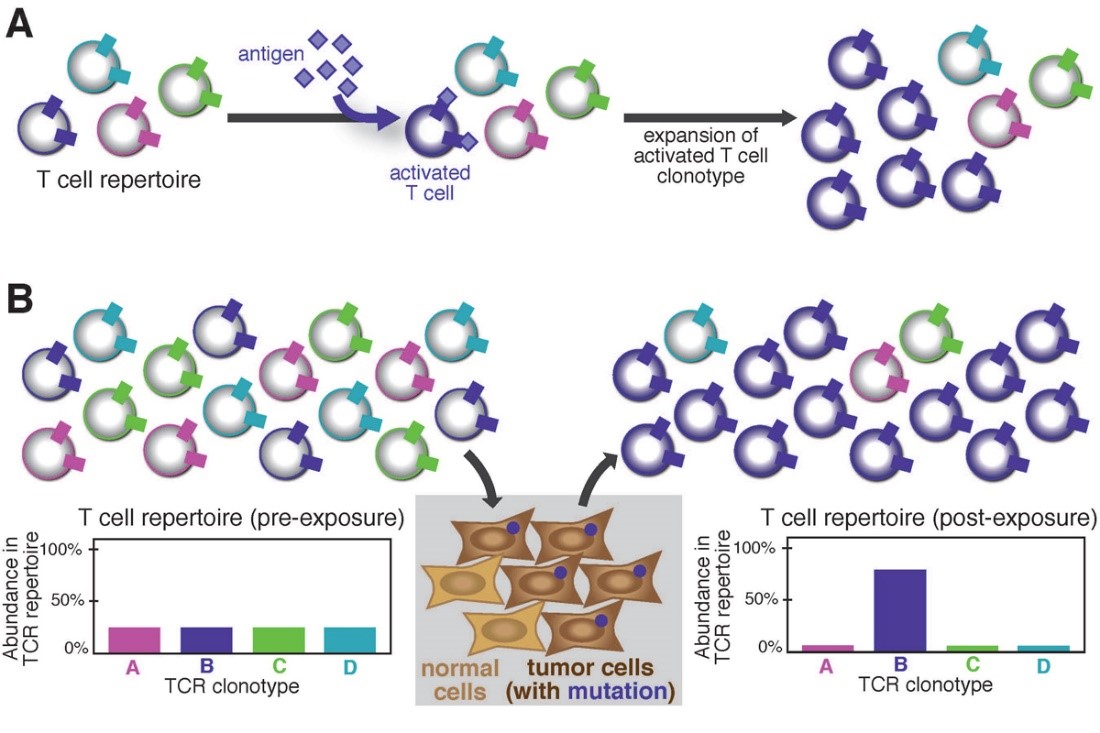
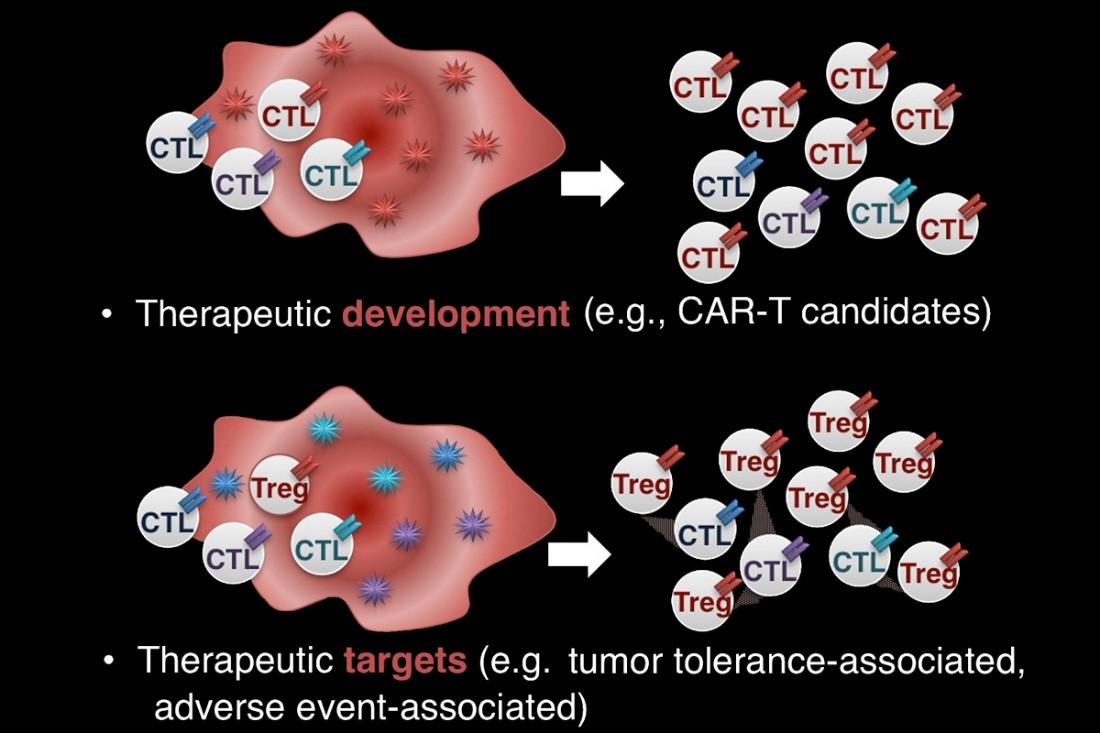
Many of these immune cells are not circulating freely in the blood, but infiltrate and provide surveillance in tissues (called tissue-infiltrating lymphocytes; TILs). Unlike the circulating population, TILs represent only a small sample of the total repertoire. T cells surveilling any tissue may be selected to reside in that particular organ or tissue based on their receptors, growth factors and other signaling molecules.
Recent advances in high-throughput next-generation sequencing let us capture the TCRs from a whole sample (using a technology called TCRseq), including both circulating blood cells and T cell-infiltrated tissue, and describe the population in terms of TCR distribution. Using statistics, we analyze the diversity of these populations, compare them to one another and look for patterns across groups of patients being treated for cancer. How does the TCR repertoire inside a tumor differ from that in the circulating blood?
We are currently defining properties that indicate tumor-specific reactivity: What does the antitumor T cell response look like when it's working? When it's failing? When it has been restored through immunotherapy? These properties may be useful as multi-dimensional biomarkers to monitor tumor progression and therapeutic response. We are also using TCR repertoire sequencing to identify receptors that could be adapted for use as antitumor therapeutics.
One of CITI's goals is to extract the immunogenomic information that will allow doctors to anticipate which patients are most likely to respond well to immunotherapy. We study tumor phenotype, or cell behavior, which is largely determined by the levels at which each gene is expressed. In particular, we use high-throughput sequencing of RNA from tumor biopsies to study how gene expression changes as cancer progresses, when therapy is given and when therapy is effective. Comparing tumors from patients who respond well to treatment versus those of patients who do respond well allows us to identify distinct tumor features that can be translated into diagnostic, prognostic and therapeutic biomarkers to be used for future patients.
The expression levels of genes also provide information about the environment in which the tumor evolves, particularly how the patient's own immune system responds. Using cutting edge computational techniques, we can integrate this information to understand what types of immune cells are successful in this antitumor immune response.
Differential expression
Tumors differ from one another, in part because each patient's immune system reacts to a tumor by using a unique set of cells to try to destroy it. Abnormal tumor cell behavior, specific antitumor immune activity, non-specific inflammatory immune activity and tissue damage all shape the gene expression profiles of both tumor and non-tumor cells in unique ways.
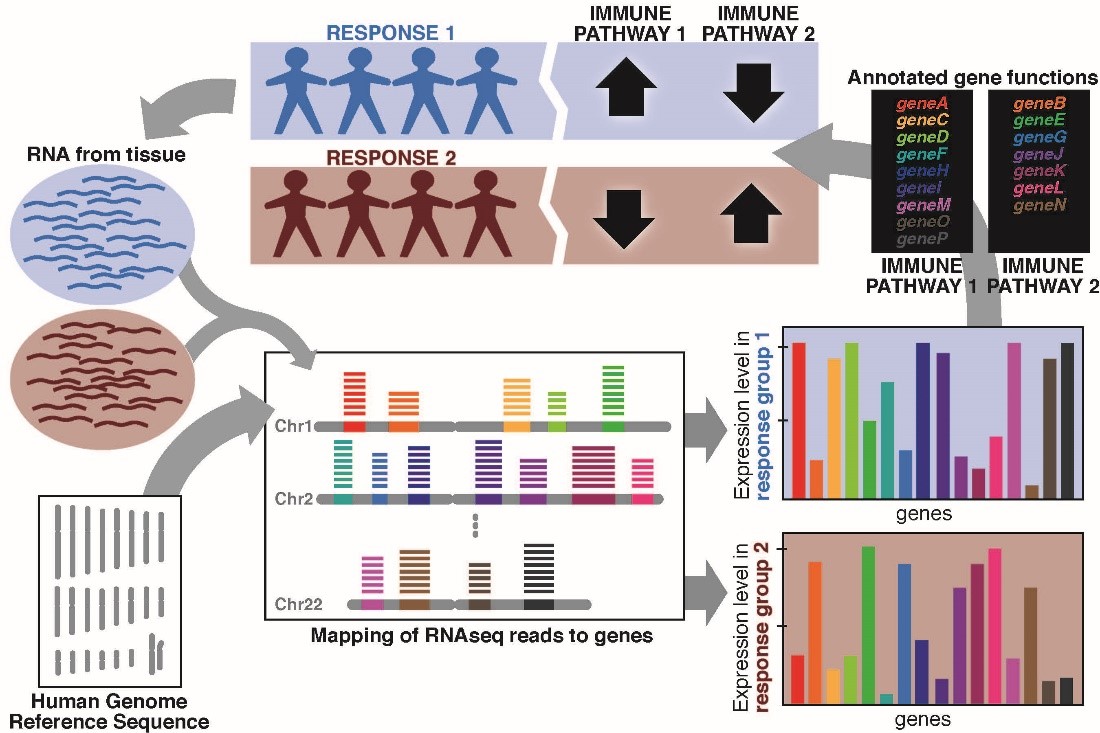
One application of differential gene expression analyses is to compare the pre-treatment and post-treatment profiles of tumors that responded to immunotherapy with those that did not. We can also identify marker genes or groups of functionally related genes that, if unusually high or low prior to treatment, correspond with better therapy response. Such predictive signatures could enable a simple pre-treatment biopsy to help tailor a patient's treatment regimen.
CITI's pipeline for automating and visualizing these analyses is constantly improving. High-dimensional data visualization tools such as oncoprints and Visne maps allow us to organize and render dozens of parameters (e.g., RNASeq gene expression data in parallel with clinical parameters) simultaneously — without sacrificing their complexity — to enrich our understanding of the cancer immune environment.
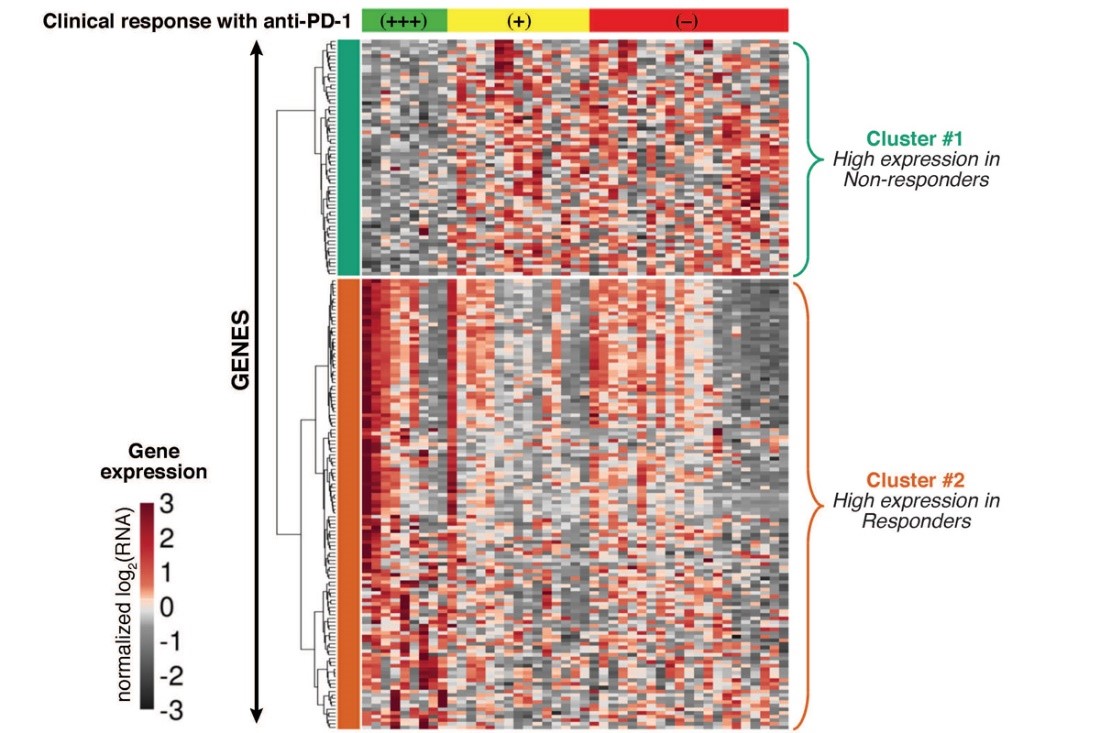
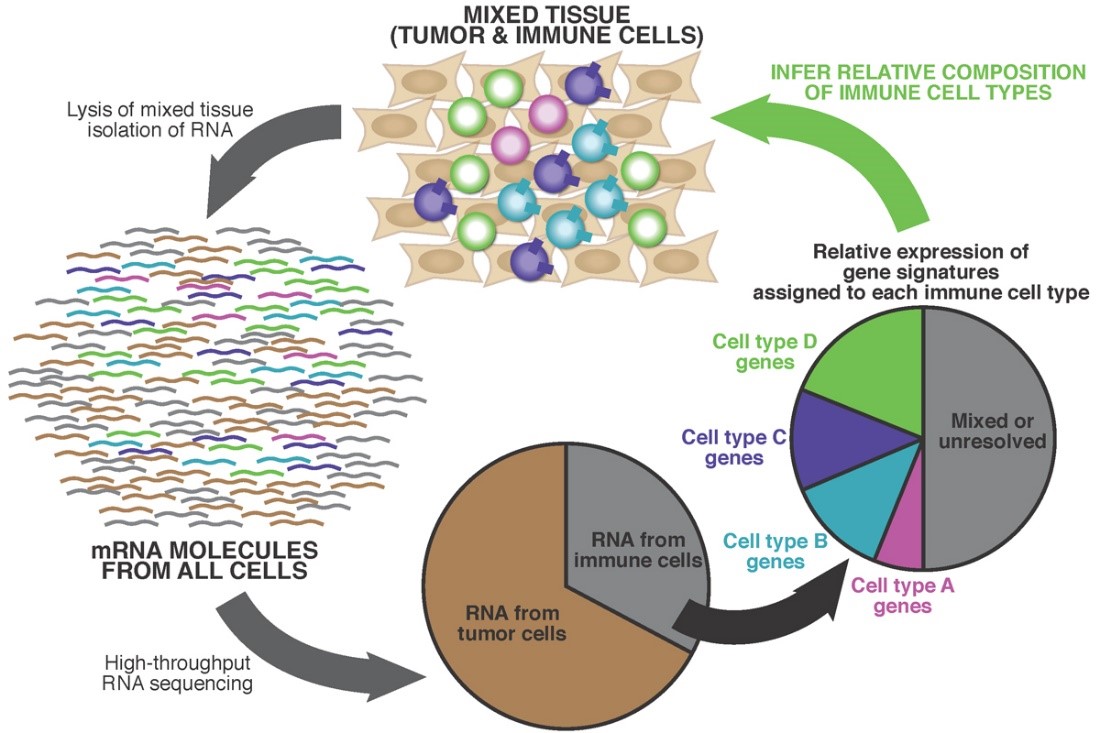
Cell composition (in silico deconvolution)
Many different immune cell types infiltrate tissues, where they perform different roles in surveilling for tumors, injuries or infections. For example, certain types of T cells are capable of directly killing dysfunctional, tumorigenic or infected cells, while monocytes and macrophages take up free-floating cell debris and present these potential antigens to T cells. This interaction, which requires both T cells and antigen-presenting cells, can help locally activate or suppress all of the T cells that recognize the same antigens. Meanwhile, B cells produce antibodies that can rapidly spread throughout the body to neutralize a particular threat. Thus, the relative abundance of different cell types can indicate which modes of tumor recognition are active, and which may be suppressed.
The type and degree of immune infiltration into tumors plays an important role in the efficacy of immunotherapy. The abundance of the messenger RNA (mRNA) of particular genes in a tissue biopsy not only allows us to identify differential gene expression between samples, but also enables us to calculate the relative abundance of different immune cell types in the local microenvironment. From the mRNA of the bulk sample, we can detect high expression of signature genes or enrichment of a subset of genes that are specific to one cell type and compare it to the expression of genes specific to other cell types. We use computational algorithms such as Supporter Vector Machines (SVMs) or Single-Sample Gene Set Enrichment (ssGSEA) to translate the expression of these signatures into relative abundances of the corresponding immune cell populations.
Because immunotherapies perform different functions—such as maintaining immune cell activation, rescuing immune cells that become exhausted or stimulating antitumor reactivity among immune cells that were previously unexposed to tumor antigens—understanding which types of immune cells are present (or not) in the tumor microenvironment has implications for predicting immunotherapy treatment response, and, therefore, choosing the right option for each patient.
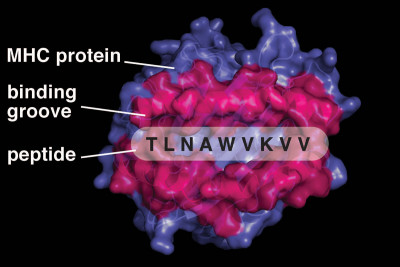
T cells recognize microbial threats and cancer by binding to degraded bits of foreign proteins (peptides) presented to them by the molecules of the major histocompatibility complex (MHC). These presentation molecules are expressed on the surface of most cell types, but appear more abundantly on certain immune cells that provide tissue surveillance.
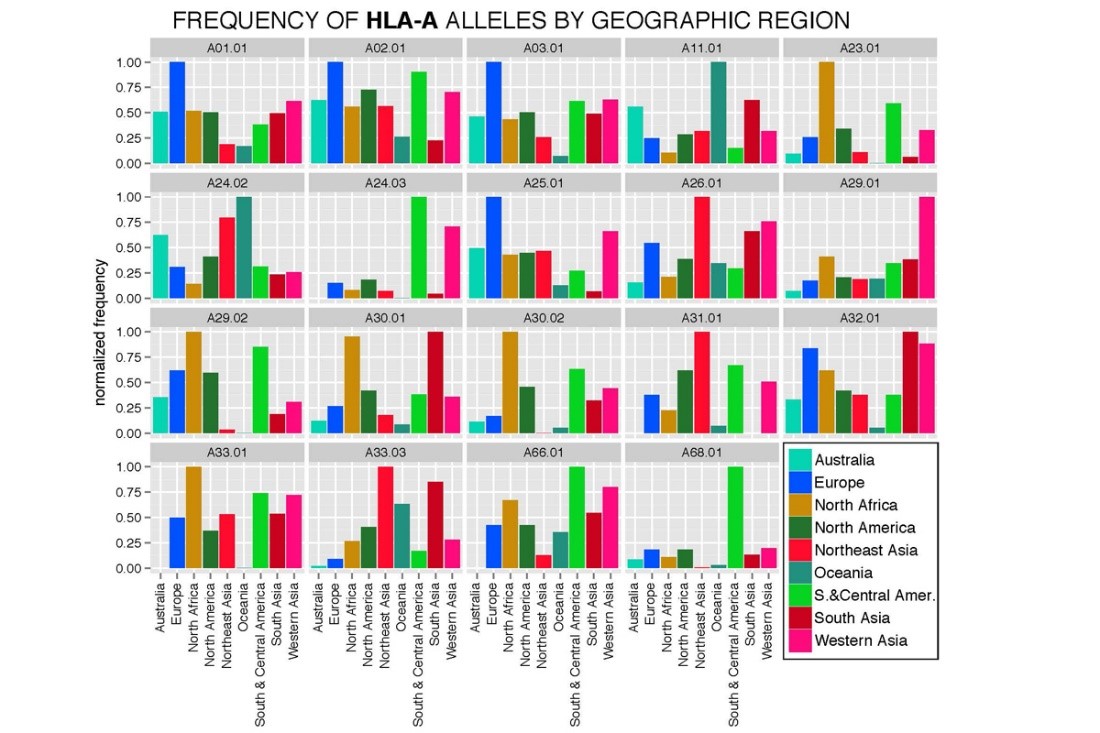
The genes that encode MHC class I proteins (called the HLA class I genes in humans) are located on chromosome 6. There are three HLA class I genes: HLA-A, HLA-B and HLA-C. Every person has two copies (alleles) of each gene (one from each parent). Since these genes are the most polymorphic (variable in DNA sequence) in the entire human genome, the six alleles each person has are often all different, and rarely do they match those of genetically unrelated individuals. There are specific alleles (e.g. HLA-A*02:01) that are more prevalent worldwide. Moreover, the frequency of HLA alleles varies across geographic regions and populations.
We examine how the HLA alleles a patient uses affects immunotherapy responsiveness. The presentation of peptides to T cells by the MHC proteins plays a critical role in the adaptive immune response and strongly influences how T cells respond. For example, some MHC molecules activate T cells strongly, which is desirable if the specific antigen represents a threat (such as a viral infection or a dysfunctional or mutated protein produced by a tumor). However, this can be dangerous if the antigen is normal and occurs on healthy cells. Because potentiating the correct recognition by T cells of self versus non-self peptides is a major function of MHCs, and this distinction becomes muddled in the case of cancer, it is important to use genomic sequencing data to identify which six HLA alleles a patient has when determining how his or her immune system will react to the mutated tumor peptides.
Currently, the gold standard for identifying which HLA alleles a patient has is PCR-based typing, in which the HLA locus is specifically amplified and then sequenced. As genomic sequencing has achieved higher coverage, in silico HLA genotyping offers an efficient alternative that is economical when a patient's genome is already being sequenced. Current software tools provide up to 99% accurate resolution for most clinical applications. However, certain clinical applications require higher accuracy; certain HLA alleles differ from their closest alleles by only a few nucleotides, so current software tools must be more precise when predicting tumor antigen presentation. As a result, we are refining the computational pipelines for HLA identification using ensemble approaches, population-based weighting, and alternative assemblies of the human reference genomes.
Understanding the cellular composition of tumor and immune cells on the level of phenotypic protein markers is a critical part of investigating tumor immunology. CITI utilizes several experimental techniques to better quantify protein expression in individual tumor and immune cells. Antibody-based flow cytometry allows for the precise quantification of extracellular and intracellular proteins of interest. Using fluorescence-activated cell sorting (FACS), individual immune or tumor cell populations can be further subdivided for downstream analysis, including DNA and RNA sequencing.
Occasionally, investigators may wish to simultaneously quantify the expression of a large number of intracellular and extracellular proteins from a single sample. Conventional flow cytometry limits the number of simultaneous parameters detectable due to fluorophore-generated spectral overlap. To overcome this barrier, CITI utilizes mass cytometry by time-of-flight (CyTOF) technology. CyTOF identifies intracellular and extracellular proteins using antibodies conjugated to rare earth heavy metals. After antibody-based staining, the sample is ionized and the antibody composition of single cells are subsequently identified. The primary advantage of CyTOF is its ability to simultaneously analyze a robust user-defined panel of cellular targets from a single sample using an antibody-based approach. Multi-parametric data can subsequently be analyzed using conventional flow cytometry software or more sophisticated techniques, including SPADE or ViSNE plots.
What is Precision Immuno-Oncology?
Precision oncology leverages the unique genetic characteristics of each patient's tumor to identify the best therapeutic options. Precision immuno-oncology takes this approach a step further to better understand:
- each patient's immune system and how it can be leveraged to control the tumor
- how the immune cells interact with tumor cells in each patient
- how a patient's cancer genome (or tumor-intrinsic features) dictates anti-tumor immunity and immunotherapeutic response.
Immunotherapies have shown remarkable success in the treatment of many cancers, including melanoma, and of the lung, colorectal, and head and neck; however, there is still a poor understanding of why certain patients do not respond well.
Precision immuno-oncology helps to answer key questions for cancer patients considering immunotherapy: who will respond best and what are the molecular mechanisms behind therapeutic response? Answering these questions will help clinicians and scientists to successfully individualize cancer care.
Immuno-oncology-based therapeutic strategies include:
- Cell Therapy
- Targeted Antibodies and ADCs
- Cancer Vaccines
- Immune System Modulators
- Checkpoint Inhibitors
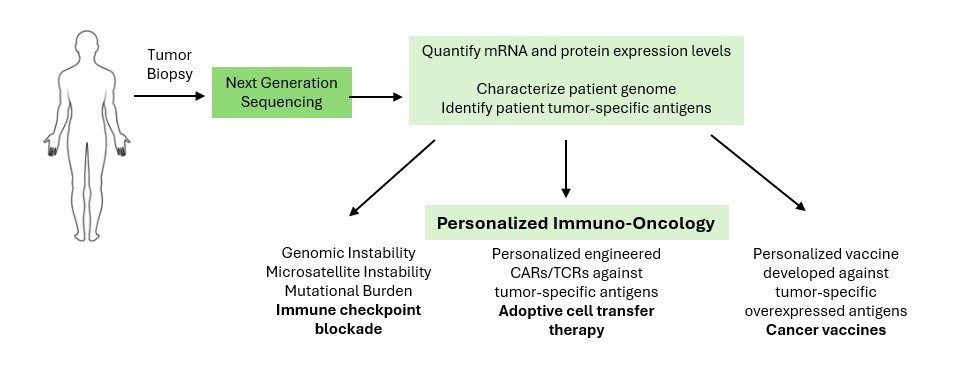
Cell Therapy & Immuno-Engineering Program
What do we do?
CITI’s Cell Therapy and Immuno-Engineering program is working to study why existing cell-based immunotherapy treatments are effective against some types of cancer, to create new therapies that target additional cancers and to expand into therapies that target other immune-related diseases. Delivering the most effective therapies requires continuous research – developing new drugs, studying them for safety and making refinements. That’s where we come in.
What makes our program unique?
The heart of the CITI Cell Therapy and Immuno-Engineering program is its translational nature, which fosters a true “bench to bedside” approach. CITI researchers collaborate with clinicians at the nearby Taussig Cancer Institute, who work to treat some of the most complex cancer cases. Our method of research allows scientists to more safely and effectively target cancers in patients receiving care just around the corner. Data generated in the lab therefore reflects the types of cancers directly affecting the community and fosters a sense of connection that is often removed from data collected in the lab. This connection helps our researchers care for their patients with the utmost attention and passion, resulting in excellent patient care.
For example, the Melenhorst Laboratory examines T cells directly from patients in the community who are undergoing immuno-gene therapies for certain types of cancer. The researchers study the relationship between these patients’ specific T cells and tumor cells, and ultimately design chimeric antigen receptors (CARs) that work to yield tangible results for patients affected by these types of cancers. Already, the CAR T-cell therapies that they have developed using this pipeline have demonstrably delivered augmented memory and endurance to the affected immune system and even durable remissions to those treated.
We are actively working to translate such conclusions to novel CAR T-cell therapies, precision engineering, and to the reduction of toxicities and cost — aspects essential to our unique approach at Cleveland Clinic.


Our Mission
Current commercial CAR T-cell products are broadly considered highly successful in a number of specific cancers.
But, we can do better than that!
CITI's cell therapy program strives to expand our reach beyond cancer treatment. The mission of CITI's Cell Therapy and Immuno-Engineering program is to translate cell-based therapy research into next-generation therapeutics. Our program seeks to empower an adaptable defense system — the immune system — to target cancer cells, the threat. Delivering the most effective therapies requires a continuous cycle of therapeutic development, clinical evaluation and further improvement. Ultimately, our goal is to develop safe, effective and affordable cell-based therapies for cancer and immune-related diseases.
What is CAR T-cell therapy?
In cases where cancers are able to evade recognition by the immune system, it has to be retrained to recognize and attack the threat. Chimeric antigen receptor (CAR) T-cell therapy is an innovative new form of personalized immunotherapy that reprograms immune cells, called T cells, to more specifically target certain types of cancer cells.
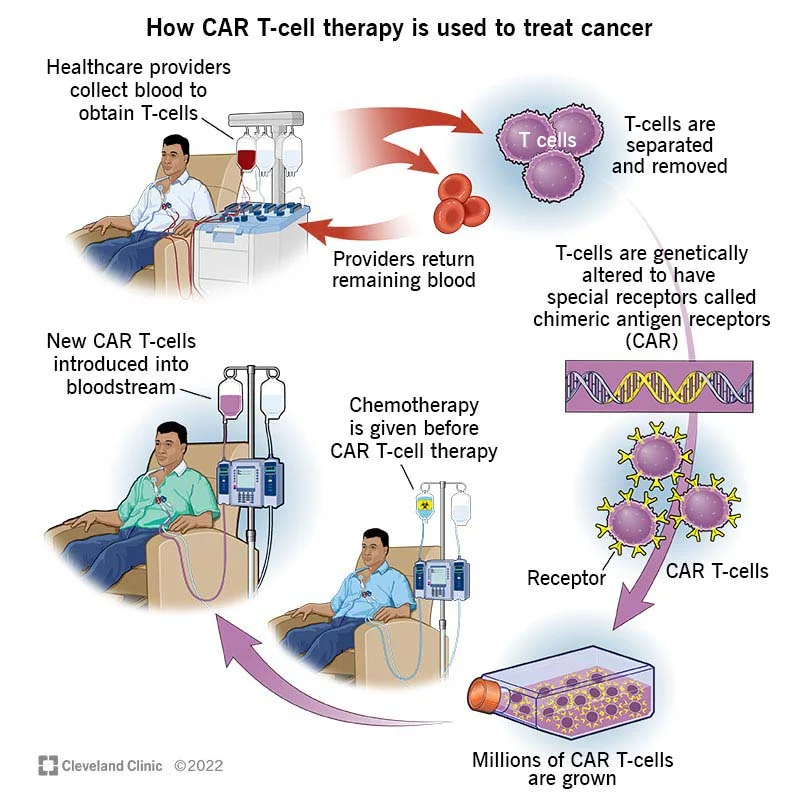
For this reprogramming to occur, patients' T cells are collected from their blood and re-engineered so they grow special structures on their surface – chimeric antigen receptors (CARs). While the CARs are grown in the lab, patients are given chemotherapy to lower their overall white blood cell count. This makes the space necessary for the newly engineered CAR T cells. After the newly engineered cells are reinjected into the patient, these receptors assist the T cells to identify and attack cancer cells throughout the patient's body. In this way, CAR T-cell therapy is personalized medicine, tailored to the patient’s specific needs.
Learn More about CAR T-Cell TherapyHighlighted Studies
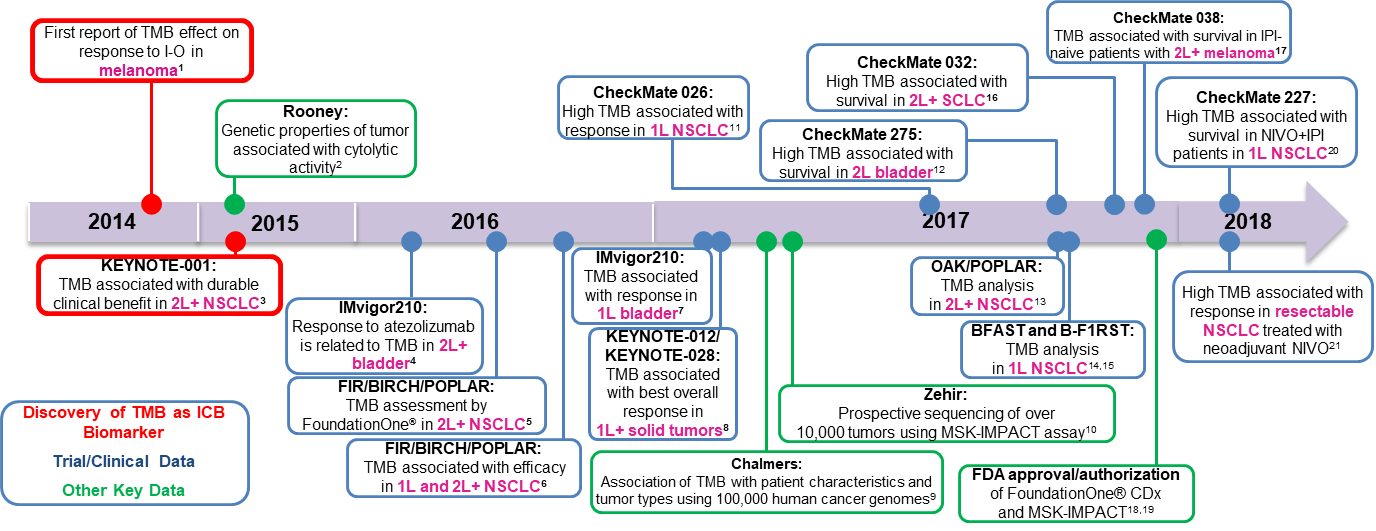
Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic
Timothy A. Chan. Mark Yarchoan, Elizabeth Jaffee, Charles Swanton, Sergio A. Quezada, Albrecht Stenzinger, Solange Peters Annals of Oncology. 2019 Jan; 30(1): 44-56.
The Chan lab discovered that mutation and neoantigen burden and MSI are primary drivers of immune checkpoint blockade therapy efficacy. The timeline of discoveries are summarized in this review. These findings sparked the FDA approval of the first tumor-agnostic approvals for cancer therapy.
View on Pubmed
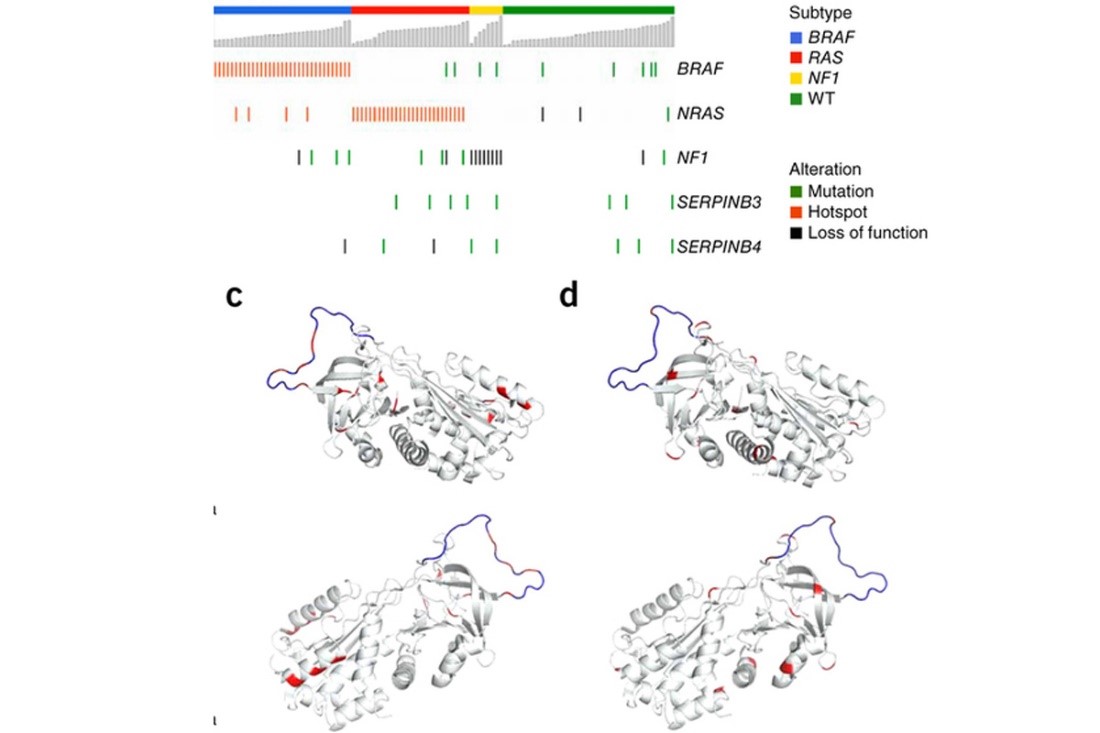
Recurrent SERPINB3 and SERPINB4 mutations in patients who respond to anti-CTLA4 immunotherapy
Nadeem Riaz, Jonathan J. Havel, Sviatoslav M. Kendall, Vladimir Makarov, Logan A. Walsh, Alexis Desrichard, Nils Weinhold, and Timothy A. Chan
Nat Genet. 2016 Nov;48(11):1327-1329.
Immune checkpoint blockade has shown significant promise as an anticancer treatment, yet the determinants of response are not completely understood. Here we show that somatic mutations in SERPINB3 and SERPINB4 are associated with survival after anti-CTLA4 immunotherapy in two independent cohorts of patients with melanoma (n = 174). Interestingly, serpins are homologs of the well-known ovalbumin antigen and are associated with autoimmunity. Our findings have implications for the personalization of immunotherapy.
View on Pubmed
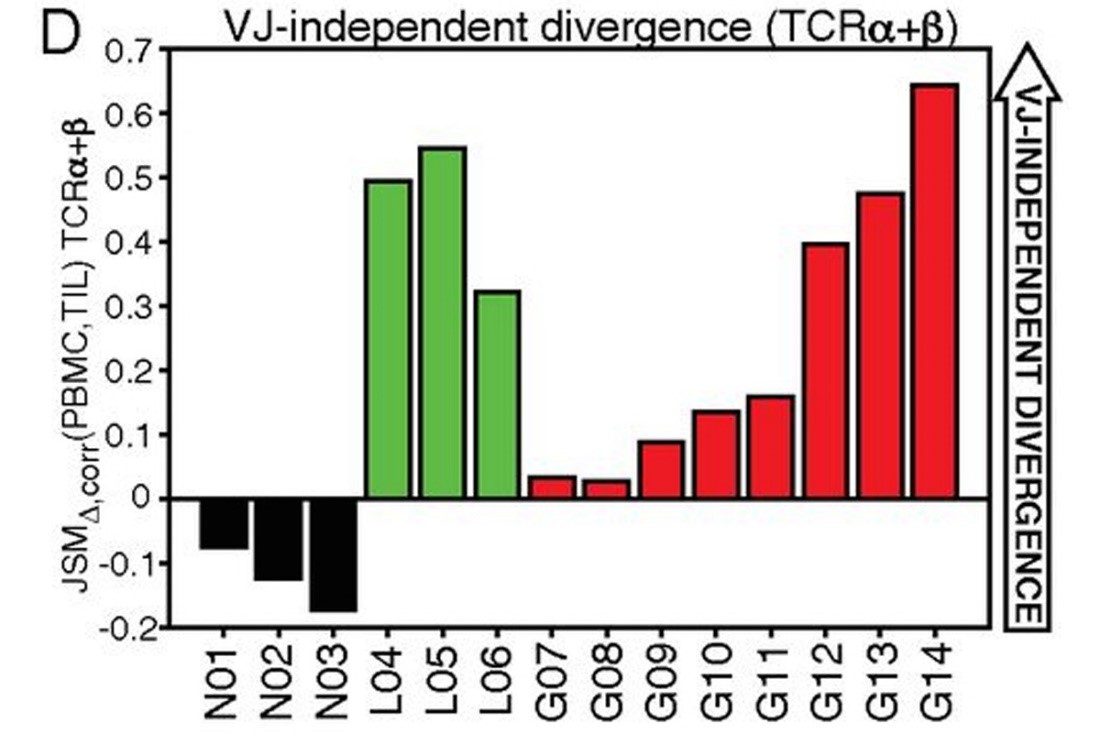
Diversity and divergence of the glioma-infiltrating T cell receptor repertoire
Jennifer S. Sims, Boris Grinshpun, Yaping Feng, Timothy H. Ung, Justin A. Neira, Jorge L. Samanamud, Peter Canoll, Yufeng Shen, Peter A. Sims, and Jeffrey N. Bruce
Proc Natl Acad Sci. 2016 Jun 21;113(25):E3529-37.
High-throughput sequencing of T cell receptor (TCR) repertoires provides a high-dimensional biomarker for monitoring the immune system. We applied this approach, measuring the extent to which the TCR repertoires of T cell populations infiltrating malignant brain tumors diverge from their peripheral blood counterparts. Our analytical strategy separates the statistical properties of the repertoire derived from VJ cassette combination usage from the VJ-independent contribution that reflects the antigen-binding component of the receptor. We discovered a TCR signature strongly inversely correlated with the VJ-independent divergence between the peripheral and tissue-infiltrating repertoires of these patients. Importantly, this signature is detectable in peripheral blood and could serve as a means of noninvasively monitoring immune response in patients.
View on Pubmed
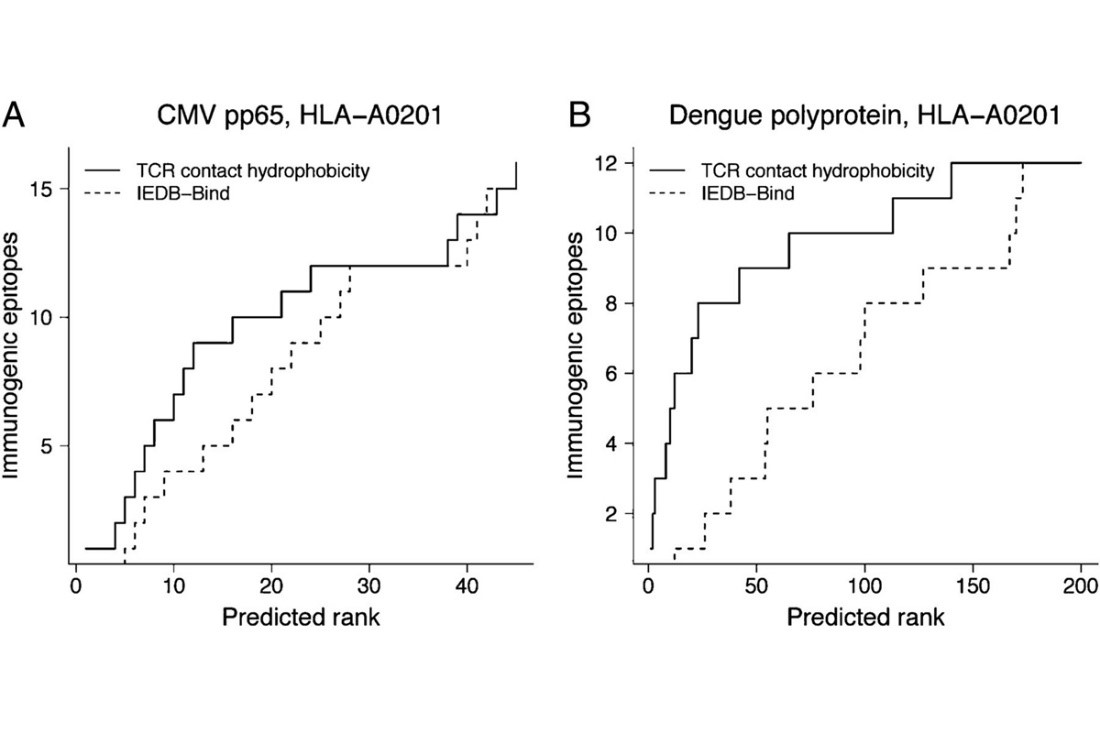
TCR contact residue hydrophobicity is a hallmark of immunogenic CD8+ T cell epitopes
Diego Chowell, Sri Krishna, Pablo D. Becker, Clément Cocita, Jack Shu, Xuefang Tan, Philip D. Greenberg, Linda S. Klavinskis, Joseph N. Blattman, and Karen S. Anderson
Proc Natl Acad Sci. 2015 Apr 7;112(14):E1754-62.
The design of effective T cell vaccines against pathogens and tumor antigens is challenged by the highly inefficient identification of the subset of peptides from a given antigen that effectively stimulate an immune response. Here we report that the relative hydrophobicity of T cell receptor contact residues is markedly enriched in immunogenic major histocompatibility complex class I epitopes in both human and murine MHCs, and in both self and pathogen-derived immunogenic epitopes. Incorporating hydrophobicity into T cell epitope prediction models increases the efficiency of epitope identification, which will reduce the time and cost of T cell vaccine development. Amino acid hydrophobicity may represent a biochemical basis by which T cells discriminate immunogenic epitopes within the background of self peptides.
View on Pubmed
Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer
Naiyer A. Rizvi, Matthew D. Hellmann, Alexandra Snyder, Pia Kvistborg, Vladimir Makarov, Jonathan J. Havel, William Lee, Jianda Yuan, Phillip Wong, Teresa S. Ho, Martin L. Miller, Natasha Rekhtman, Andre L. Moreira, Fawzia Ibrahim, Cameron Bruggeman, Billel Gasmi, Roberta Zappasodi, Yuka Maeda, Chris Sander, Edward B. Garon, Taha Merghoub, Jedd D. Wolchok, Ton N. Schumacher, and Timothy A. Chan
Science. 2015 Apr 3;348(6230):124-8
Checkpoint blockade immunotherapy provides stronger benefits in some patients than in others. This study involves the use of pembrolizumab, an antibody which blocks the programmed cell death-1 (PD-1) "exhaustion" receptor on T cells, to treat non-small cell lung cancer (NSCLC). The findings demonstrate that the number of protein-altering mutations (and higher predicted neo-antigen load) in the tumor correlates with objective response, durable clinical benefit, and progression-free survival. Signature types of nucleotide mutations and mutations in DNA repair pathways were also enriched among anti-PD-1 responders. This study also highlights the neo-antigen specific CD8+ T cell responses in one particular patient; these responses coincided with tumor regression, providing a circumstantial mechanistic link between the anti–PD-1 therapy and the antitumor effects of neo-antigen-specific T cells.
View on Pubmed
Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade
Nicholas McGranahan, Andrew J. S. Furness, Rachel Rosenthal, Sofie Ramskov, Rikke Lyngaa, Sunil Kumar Saini, Mariam Jamal-Hanjani, Gareth A. Wilson, Nicolai J. Birkbak, Crispin T. Hiley, Thomas B. K. Watkins, Seema Shafi, Nirupa Murugaesu, Richard Mitter, Ayse U. Akarca, Joseph Linares, Teresa Marafioti, Jake Y. Henry, Eliezer M. Van Allen, Diana Miao, Bastian Schilling, Dirk Schadendorf, Levi A. Garraway, Vladimir Makarov, Naiyer A. Rizvi, Alexandra Snyder, Matthew D. Hellmann, Taha Merghoub, Jedd D. Wolchok, Sachet A. Shukla, Catherine J. Wu, Karl S. Peggs, Timothy A. Chan, Sine R. Hadrup, Sergio A. Quezada, and Charles Swanton
Science. 2016 Mar 25;351(6280):1463-9
Antitumor immunity is influenced by the repertoire of neo-antigens created by somatic mutations within a tumor. Spatial heterogeneity, in other words – the diverse concentration of tumor mutational profiles within the tumor microenvironment– can highly impact the degree to which the immune system can recognize the neo-antigens. Some neo-antigens are present in virtually all tumor cells (clonal), while others are present only in a fraction of cells (subclonal). This study demonstrates that enrichment for clonal neo-antigens is associated with an improved clinical outcome with checkpoint blockade immunotherapy, and increased tumor infiltrating lymphocytes.
View on Pubmed
Genetic basis for clinical response to CTLA-4 blockade in melanoma
Alexandra Snyder, Vladimir Makarov, Taha Merghoub, Jianda Yuan, Jesse M. Zaretsky, Alexis Desrichard, Logan A. Walsh, Michael A. Postow, Phillip Wong, Teresa S. Ho, Travis J. Hollmann, Cameron Bruggeman, Kasthuri Kannan, Yanyun Li, Ceyhan Elipenahli, Cailian Liu, Christopher T. Harbison, Lisu Wang, Antoni Ribas, Jedd D. Wolchok, and Timothy A. Chan
N Engl J Med. 2014 Dec 4;371(23):2189-99
Higher numbers of mutations, leading to increased numbers of neo-antigens, may be recognized by the immune system as foreign. This study was the first to demonstrate — in human patients — that tumors with increased mutational loads are indeed associated with improved outcomes after immunotherapy. Furthermore, T cells were identified in several patients that responded to the neo-peptides predicted by our computational pipeline, but not to their respective wild-type peptides. Many different types of antigens — such as cancer-testis antigens, viral antigens, and neo-antigens — could lead to immune recognition of a tumor as foreign; however, this study provided the first evidence from patients that suggests neo-antigens' critical role in this process. At present, the clinical evidence for other classes of antigens remains poor. The commonality of certain features of the tumor-associated neo-peptides (e.g. homology to known antigens or particular amino acid motifs) suggested that these features may be helpful in identifying patients who would respond strongly to therapy.
View on Pubmed
U54 Gen-Rad ROBIN Center
About
Multi-principal investigators Dr. Timothy Chan of the Cleveland Clinic Lerner Research Institute and Dr. David Yu of Emory University’s School of Medicine were awarded a Radiation Oncology-Biology Integration Network (ROBIN) U54 grant from the National Cancer Institute for a consortium study entitled “Genomic and Microenvironmental Determinants, Temporal Dynamics, and Efficacy of Radiation-Based Combination Therapies.” They will work together to study radiotherapy in combination with antibody-drug conjugates and immune checkpoint inhibitors to observe how combined radiation and immunotherapy treatments may be used to treat bladder cancer and head and neck cancers.
Learn more about the U54 Gen-Rad ROBIN Center from Dr. Timothy Chan on the Cleveland Clinic's Cancer Advances Podcast.
Gen-Rad Center Overview
The Gen-Rad center focuses on elucidating two factors underlying the efficacy of radiation-based combination therapies: genomic and microenvironmental determinants, and temporal dynamics. Two innovative trials are underway. The first trial interrogates the effects of radiation plus antibody-drug conjugates as a therapy for bladder preservation in advanced stage bladder cancer, and the second trial investigates the effects of radiation plus immunotherapy in head and neck cancer. The biospecimens derived from the trials will synergize with three laboratory-based research projects that aim to optimize multimodal, radiation-based definitive treatment strategies. Three cores ranging from administration, career development, and biospecimen banking will harmonize the inter-institutional translational projects.

Timothy Chan, MD, PhD
Chair, Center for Immunotherapy & Precision Immuno-Oncology
Lab Profile

David Yu, MD, PhD
Chair of Cancer Biology/Associate Professor, Department of Radiation Oncology
Emory University
Our aims:
- To understand the molecular mechanisms that underlie the efficacy of combination treatment with radiation plus antibody drug conjugates.
- To improve the identification of patients who are sensitive or resistant to RT-based therapies based on new insights into transcriptional dynamics and temporal reprogramming during treatment with radiation-based therapies.
- To identify the differential mechanisms underlying the anti-tumor activities of radiation plus cisplatin versus radiation plus immune checkpoint blockades.
For more information, please contact Christine Lee-Poturalski ([email protected]), CC-Emory ROBIN Center Coordinator, or Timothy Chan ([email protected]), Chair of CITI.
Follow @NCITreatment on Twitter for updates on the ROBIN Centers.
Learn More About NCI U54 ROBIN Centers